新闻网讯(通讯员高妍)物质和能源构建了现代生活,而传统的物质转化通常需要以化石能源为基础,带来了环境污染等问题。雷爱文教授团队最近以绿色能源——光或电为能量输入,实现了高值化合物的合成,取得了系列重要科学进展,既为绿能转化提供新途径,也为物质转化提供新手段。相关研究成果近日连续在NatureCatal.,JACS,ACIE发表。
7月22日,Nature Catalysis《自然·催化》在线发表了雷爱文课题组在三级烷基烯丙胺合成方向的最新研究成果,论文题为“Site-selective amination towards tertiary aliphatic allylamines”(位点选择性胺化反应构建三级烷基烯丙胺)。高研院博士后王盛淳和博士研究生高一鸣为共同第一作者,武汉大学雷爱文教授和戚孝天教授为论文的共同通讯作者,高研院为论文的第一署名单位。

烷基烯丙胺是基本有机骨架,广泛存在于药物分子中。利用烷基胺和廉价烯烃构建烷基烯丙胺是一种理想方式,但却困难重重:(1)烷基胺具有较强配位性,容易导致催化剂失活;(2)三级烷基烯丙胺具有较强的氧化还原活性,产物容易氧化变质;(3)理想的氧化析氢偶联方式热力学不利;(4)自由基加成中间体双键重构选择性难以控制。
本文利用钴肟催化剂的氢原子攫取能力(HAT),实现了双键的位点选择性转移,解决了位点选择性难以控制的难题;同时释放氢气,避免了了烯丙胺合成中底物预活化步骤以及当量氧化剂的使用,原子经济性显著提高(图1)。
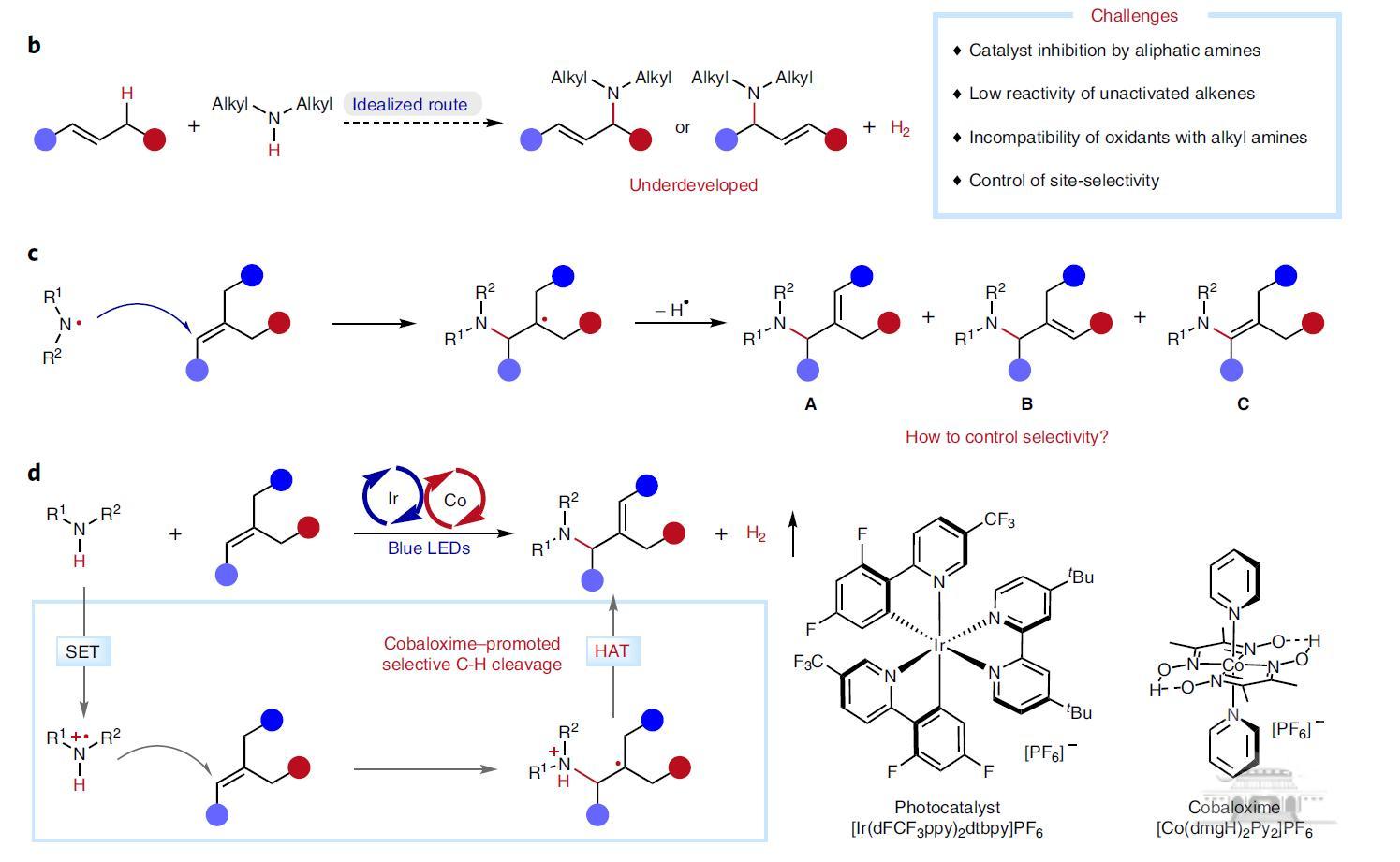
图1 烷基烯丙胺合成中的挑战与该策略(来源:Nature Catalysis)
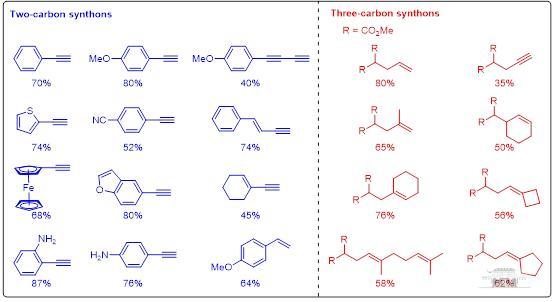
超过90个底物拓展证明该策略具有底物适用性广、原子经济性高、位点选择性优异等优势,并适用于抗菌药的合成,还可以拓展到胺化柠檬烯的精准合成中(图2)。作者通过XAFS,EPR等技术证明了反应经历了自由基反应历程,戚孝天教授团队详细的DFT计算证明了反应可能经历了氮自由基阳离子反应历程。
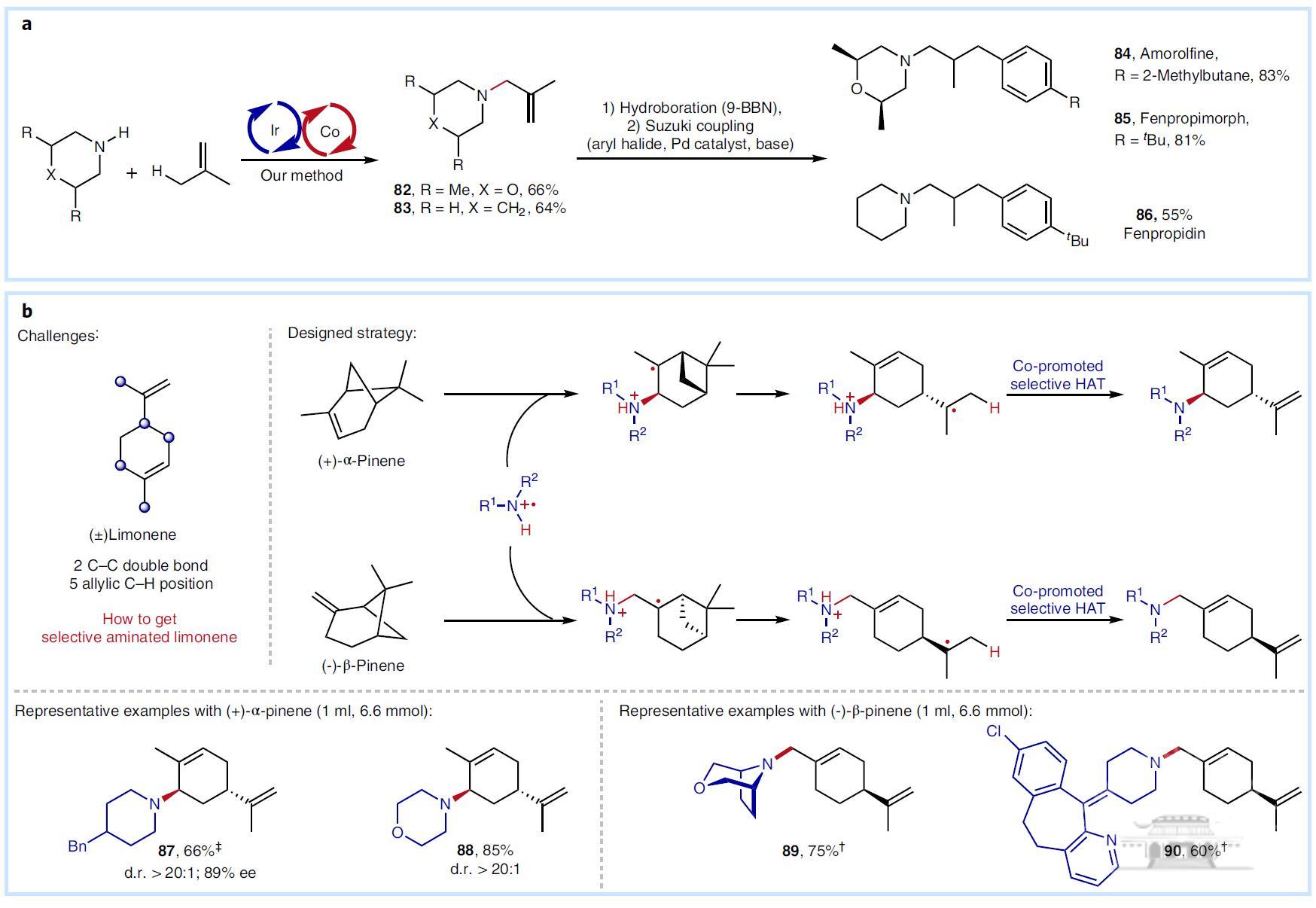
图2 抗菌药合成与萜烯的精准转化
综上,利用光催化产生氮自由基阳离子与钴催化HAT过程协同的策略,实现了烷基烯丙胺的精确合成。该策略选择性源于钴肟催化剂独特的HAT模式,优先攫取位阻小的烯丙位C-H键,从而为该反应提供了优异的选择性。该策略不仅克服了烷基烯丙胺合成的长期挑战,并助力了合成化学中对烯烃烯丙位选择性转化的长期探索。
该研究得到了国家自然科学基金项目(22031008)和武汉市自然科学基金项目(2020010601012192)的支持。
文章链接:https://www.nature.com/articles/s41929-022-00818-y
7月21日,Journal of the American Chemical Society《美国化学会会志》在线发表了雷爱文课题组在环状烷基醇碳碳断键官能团化方面的最新研究成果,论文题为“ElectrophotochemicalCe-Catalyzed Ring-Opening Functionalization of Cycloalkanols under Redox-Neutral Condition: Scope and Mechanism”(光、电、铈协同催化环状醇开环官能团化:应用及机理)。高研院博士研究生杨招凉和博士后杨大力为共同第一作者,雷爱文教授和黄志良博士为论文的共同通讯作者,高研院为论文的第一署名单位。
醇类化合物在糖类和药物中广泛存在。在合成方面,烷基醇的选择性官能团化一直是合成化学家们不断追求的目标。碳碳键是构成有机化合物的基本骨架,选择性的切断碳碳键来重新构筑更加具有价值的碳碳和碳杂键也一直备受关注。从环状烷基醇出发,产生烷氧自由基,从而为切断碳碳键提供了很好的途径。以往产生烷氧自由基的方法都因其特殊性,大大限制了反应的类型和官能团的耐受性。与以往产生烷氧自由基的方式相比,雷爱文课题组利用廉价铈金属催化,采用光电结合的方式,不需要强氧化剂和还原剂的存在,在氧化还原中性的条件下就能够很好的直接从羟基出发产生烷氧自由基。
该方法在温和条件下不仅实现了不同环张力的开环,并且能够实现多种多样的官能团化,从而大大的拓展了该方法的实用性(图3)。

图3 开环不同官能团化
在机理研究方面,作者通过同步辐射,第一次在线捕获到金属铈催化剂在反应过程中的Ce(III)和Ce(IV)价态变化,证实了电氧化的作用。接着作者利用电子顺磁共振波谱(EPR),利用DMPO作为自由基捕获剂,成功证实了反应过程中是产生氧自由基,从而实现开环官能团化的过程 (图4)。
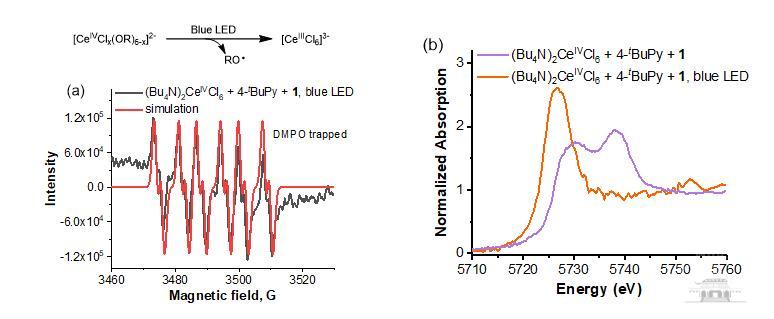
图4 机理研究
综上,该课题组利用光电结合结合,廉价金属铈催化,通过配体到金属电荷转移(LMCT)策略,实现了温和条件下不同环状烷基醇的开环官能团化。该策略为廉价金属在电化学转化过程中提供了更为广泛的运用,并为烷基醇的转化提供了全新的思路。
该研究得到了国家自然科学基金项目(22031008)、武汉市自然科学基金项目(2020010601012192)的支持。
文章链接:https://pubs.acs.org/doi/abs/10.1021/jacs.2c05520
近日,《德国应用化学》(Angew. Chem. Int. Ed.)在线发表雷爱文的最新研究成果,论文题为“Synthesis of Cyclopentene Derivatives via Electrochemically Induced Intermolecular Selective (3+2) Annulation”(电化学诱导分子间选择性(3+2)环化构建环戊烯衍生物)。高研院博士后关志朋和博士研究生朱书祥为共同第一作者,武汉大学雷爱文教授、张恒副教授和黄志良博士为论文的共同通讯作者,高研院为论文的第一署名单位(图5)。
图5 电化学诱导分子间选择性(3+2)环化构建环戊烯衍生物
环戊烯是许多天然产物、生物活性和功能分子中常见的核心骨架,也是复杂分子合成中重要的合成砌块。由于环戊烯化合物的高附价值,许多化学家致力于探索合成环戊烯有效的合成路线。例如,Michael卡宾插入反应、Conia-ene反应、乙烯基环丙烷重排反应、过渡金属催化的环化反应以及膦催化的烯丙酸酯与缺电子烯烃的[3+2]环化等是合成环戊烯的有效方法。此外,研究者们通过使用C-X/C键断裂作为碳自由基前体,开发了一系列自由基介导的(3+2)环化加成反应构建环戊烯。尽管上述方法取得了很大进展,但是直接使用C-H化合物作为碳自由基源,通过(3+2)环化合成多官能团化的环戊烯是一种未被开发的绿色合成方法。
近年来,电化学的复兴给自由基反应带来了新的活力,同时mediator在电解中起着至关重要的作用,可以提高催化效率,控制反应选择性,避免底物和产物的过度氧化或还原。在此基础上,团队以烯丙基丙二酸酯作为自由基前体以及三碳合成子,炔烃作为二碳合成子,在电化学条件下实现了mediator介导的(3+2)自由基环化,构建一系列环戊烯衍生物(图6)。
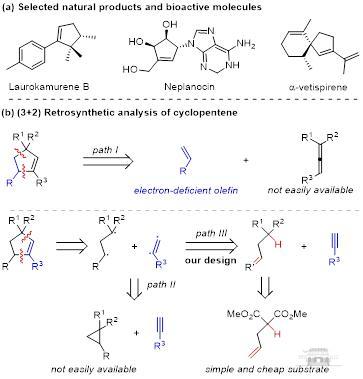
图6 环戊烯衍生物逆合成分析
在最优条件下,作者首先对炔烃的适用范围进行了考察(图7)。在NaI作为mediator和碘源的条件下,反应对于芳基/杂环炔烃,烯炔,联炔以及含有药物片段的炔烃都适用,得到一系列碘代环戊烯。对于三碳合成子,作者也进行了考察,含多个取代基的底物都可以兼容。值得注意的是,当使用烯烃代替炔烃作为二碳合成子,还可以得到环戊烷衍生物。当使用二茂铁作为mediator的条件下,得到了一系列氢化环戊烯的结构。放大实验以及衍生化实验证明了该方法的潜在应用价值。
图7 部分底物拓展
在该工作中,团队首次利用mediator介导的电化学方法实现了分子间(3+2)环化,构建了一系列高附价值的环戊烯/烷衍生物。该方法以商业可得的原料作为底物,避免了不稳定或难以制备底物的使用。同时,控制实验、DFT计算和循环伏安实验表明该途径可能涉及SET过程;高区域选择性是由动力学控制的。
该研究得到了国家自然科学基金项目(22031008)、国家重点研发计划(No.2021YFA1500104)、中国博士后科学基金(2021M702516)和武汉市科学基金(2020010601012192)的支持。
文章链接:https://doi.org/10.1002/anie.202207059
据了解,雷爱文深耕绿色氧化偶联十五载,以发展绿色和可持续发展的现代有机合成化学作为核心思想,提出了一种无牺牲性氧化剂实现脱氢反应的通用策略:放氢气氧化交叉偶联,此策略摒弃了传统方法中当量氧化剂的使用,避免了从原料到产物转化过程中氧化剂的干扰。利用绿色电能驱动化学反应,该策略可应用于大规模绿氢制备、有机膦与硫化学品等高值化工品的高效生产。放氢气氧化交叉偶联为传统化学品的合成提供一条绿色途径,也为绿色能源转化提供了新的应用策略。迄今为止发表论文450余篇,在JACS(32篇)、ACIE(48篇)、NatureCatal.(2篇)、Nat.Commun. (16篇)、Chem(2篇) 、Chem. Rev.(4篇)、Chem. Soc. Rev.(3篇)、Acc.Chem. Res.(2篇)等影响因子大于12 的杂志上发表研究论文130余篇,总被引用超29000余次,H 因子为96。2016-2022年连续入选Thomson Reuters和Elsevier的全球“高被引科学家”。
雷爱文课题组网站:http://aiwenlei.whu.edu.cn/
以上内容由大学时代综合整理自互联网,实际情况请以官方资料为准。

|
 |